 Tianqi Tu
Tianqi Tu Zhenghong Peng3†
Zhenghong Peng3† Hongqi Zhang
Hongqi Zhang- 1Department of Neurosurgery, Xuanwu Hospital, Capital Medical University, Beijing, China
- 2China International Neuroscience Institute (China-INI), Xuanwu Hospital, Capital Medical University, Beijing, China
- 3Health Management Department, The Affiliated Hospital of Southwest Medical University, Luzhou, China
Cerebral cavernous malformation (CCM) is a type of vascular anomaly that arises due to the dyshomeostasis of brain capillary networks. In the past two decades, many advances have been made in this research field. Notably, as a more reasonable current view, the CCM lesions should be attributed to the results of a great number of additional events related to the homeostasis disorder of the endothelial cell. Indeed, one of the most fascinating concerns in the research field is the inflammatory perturbation in the immune microenvironment, which would affect the disease progression as well as the patients’ outcomes. In this work, we focused on this topic, and underlined the immune-related factors’ contribution to the CCM pathologic progression.
General Description of Cerebral Cavernous Malformation (CCM)
Cerebral cavernous malformation (CCM) is a type of vascular anomaly that arises due to the defects of brain capillary networks in the central nervous system (CNS). Familial or sporadic gene mutations are widely accepted as the molecular basis of CCM (1–3). Tightly packed by the fragile vessel walls, these lesions exist dynamically. The weakness of tight junction connection and the endothelial proliferation are considered to be the main pathologies (4–6). As a consequence, flowing blood is prone to leak into the surrounding tissue through these abnormal structures. Rupture and acute bleeding can be more serious results combined with a range of complications (7–10). Evidence has described that familial and sporadic CCM patients suffered annual rupture risks of 4.3-6.5% and 0.4-3.1%, respectively, which occurred in the cerebral parenchyma, although rarely resulted in subarachnoid hemorrhage (11). Meanwhile, hemorrhagic stroke is the primary cause of disability in patients. All CCMs are thought to harbor occult bleeding, as MRI revealed that CCMs were often accompanied by hemosiderin halo signals (11–14). Moreover, although different clinical studies documented various annual symptomatic hemorrhage rates, natural history studies have identified that, both in familial and sporadic patients, past hemorrhage history is a significant risk factor for rebleeding and subsequent poor sequalae (12–16).
Although it is unlikely to become impossibly huge in the short-term, like tumors, this benign vascular disease could cause a series of neurological symptoms, including seizures, which is one of the most common reasons for hospital visit, non-specific headaches, and even neurological sensory and motor function deficits (17, 18). Once these symptoms appear, the life quality of patients can be greatly affected (7, 9, 15, 18).
For decades, progressive understanding of this vascular anomaly’s pathogenesis has been reported from clinical and basic research (19). Notably, the current view of the CCMs should be regarded as the results of the intersection of numerous factors, such as immune, hemodynamics, and angiogenesis, which formed a complex network of the disease pathology (20). Thus, we reviewed the major breakthroughs in this field in recent years and focused on the potential mechanisms between immune and the whole disease process, involving occurrence, progression, and complications, aiming to provide promising ideas for the exploration of clinical trials and therapeutic targets.
Genetic Mutations Are the Molecular Basis
To date, it is widely accepted that the initial triggers of both familial and sporadic CCM formation have been attributed to genetic mutations. Benefited from advances in multidisciplinary development and technological innovation, mutated genes can be more comprehensively supplemented. In line with this, the signaling pathways’ regulation from the molecular level could also be summarized in a more detailed state.
Genetic Mutations and CCM Pathogenesis
CCM diseases can be broadly divided into sporadic and familial diseases. Sporadic CCMs, which accounted for the majority (80~ 85%), usually presented as isolated lesions. Familial CCMs (15~20%) were followed by an autosomal dominant inheritance pattern and usually presented with multiple lesions (1, 19, 21, 22). The loss of function (LOF) mutations of CCM1 (KRIT1, Krev interaction trapped protein 1) (23–27), CCM2 (MGC6407, encoding a protein named malcavernin) (28–33), and CCM3 (PDCD10, Programmed cell death protein 10) (34–36) have been thought to be the culprits of familial CCM. Once the LOF germline mutation occurs and leads to endothelial cell dysfunction, it may cause the occurrence of CCMs. Recently, the ‘two-hit’ hypothesis in CCM pathophysiology has been strongly suggested to explain some clinical phenomena. Accordingly, the ‘first hit’ could be conceptualized as germline or somatic mutation causing one allele loss in all cells. And the ‘second hit’ could be the other allele’s somatic mutation occurring in just some cells, which can happen at an indeterminate point and trigger the initiation of the lesions. However, accumulated evidence in animal models clearly demonstrated that the homozygous deletion of a CCM gene is not sufficient to cause CCM disease, leading us to think that additional determinants are required for the lesion occurrence, including immunologic and inflammatory events.
In one of our recent works, we first revealed the novel somatic activation mutations, also called gain of function (GOF) mutation, in the MAP3K3 and PIK3CA genes in most sporadic CMs (cavernous malformations) (1). Through whole-exon sequencing (WES) and ddPCR (droplet digital PCR) detection, we identified a total of 73 sporadic patients with somatic missense variants in MAP3K3 (NM_002401.3, c.1323C>G, p.Ile441Met) and PIK3CA (NM_006218.2, in exon 7:c.1258T>C,p.Cys420Arg; in exon 9: c.1624G>A, p.Glu542Lys, c.1633G>A, p.Glu545Lys; and in exon 20:c.3140A>Gp.His1047Arg) in our 81 patients’ cohort(90.1%). Interestingly, superposition mutations of both MAP3K3 and PIK3CA were also found in 14 (19.2%) of the 73 sporadic patients. These results have also been demonstrated in recently published studies (21, 37), which undoubtedly opened a new chapter for the CCM research field. However, considering the nature of sporadic CM single lesions and its non-genetic tendency, and the low mutation detection rate in previous studies, the existence of other undiscovered somatic mutations cannot be ruled out.
Molecular Pathology and Signaling Pathway Mechanism
In general, the pathological features would reveal the essence of disease, including abnormality of endothelial cell tight junctions, proliferation of glia and endothelial cells, and even the alteration in the cytoskeleton and cell volume (4, 22, 35). Typically, CCM lesions are mulberula-like clusters of enlarged endovascular lumens. Surrounded by segmental layered basal membrane and gliotic tissue, there is little or no infiltration into the brain parenchyma (Figure 1). An impaired endothelial barrier would facilitate the infiltration of immune cells, which would potentially accelerate the lesion’s progression by further stimulating angiogenesis and gliogenesis (20).
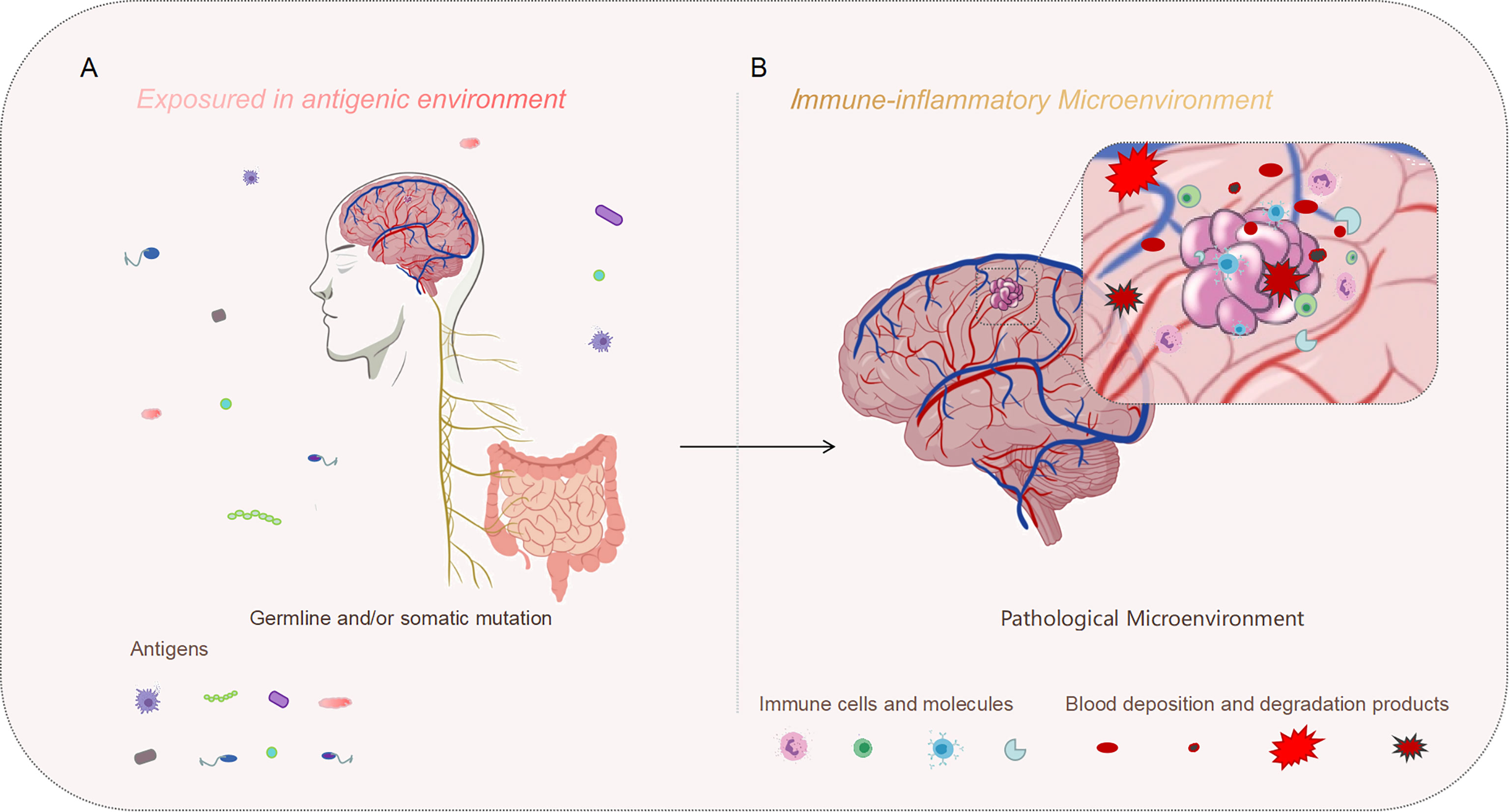
Figure 1 Genetic mutation might be the initial motivation, and the inflammatory perturbation in the immune microenvironment may drive the progression of the disease. (A) On the macro level, our bodies are constantly exposed to an antigenic environment, which could be one of the synergistic agents leading to CCM formation. (B) On the micro level, CCM lesions exist in the immune microenvironment. The immune microenvironment characterized by inflammation may aggravate the development of the typical pathology, meanwhile, repeated microbleeds could also exacerbate the inflammatory infiltration of this pathological background.
Molecular signaling pathways might be the focus of non-surgical management studies, because once the pharmacological interventions work, they may be tied to these molecular targets (19, 38). Thorough summaries about signaling pathways have been made in recent works, and the classic pathways and various upstream inputs and downstream effectors implicated in CCM signaling have also been involved (19). This shall not be repeated in too much detail in this work, but several points should be noted. Firstly, since the signaling pathways converged in a large network, changes in any one molecule can affect the entire architecture. Thus, a better understanding of CCM pathomechanism would be based on a comprehensive perspective, instead of the one - sided tip. Secondly, distinct mutation-related molecular pathways may feature differently in the pathological process (19). Generally, MEKK3 (mitogen-activated protein kinase kinase kinase 3) activation might be one of the core targets, which could help to explain why both the LOF mutation in CCM genes and the GOF mutation in MAP3K3 are sufficient to initiate CCM formation (32, 39–41).
However, unlike CCM complex or MAP3K3 mutation, the PIK3CA mutation has been shown to be associated with the proliferation of lesions. PIK3CA mutation was previously reported for venous and lymphatic malformations (42, 43), and PI3K/Akt/mTOR pathway is a critical downstream target which exacerbates the lesion growth (4, 44). Strong synergy was observed when both CCM complex LOF mutations and PIK3CA GOF mutations co-existed (4). Thirdly, inflammatory signal is one of the major inputs in endothelial cells that affected CCM disease pathogenesis. As TLR4 (toll-like receptor 4) receptors and downstream MEKK3-KLF2/4 signaling activated, the growth of CCM lesions in the brain were observed to accelerate (45). These unexpected findings have reminded us that non-genetic factors involved in the immune responses and inflammatory contexts could also play important roles in regulating these molecular networks (45, 46).
Correlations Between Genotypes and Phenotypes
Different genotypes of mutations may be important factors leading to different clinical phenotypes. In familial CCMs, different mutated genes are thought to be associated with several clinical features. It was reported that there is a certain correlation between KRIT1 mutation and cutaneous venous malformations (47–50). Individuals with CCM2 mutations were more likely to be asymptomatic and have fewer lesions (51–53). Meanwhile, due to the significant roles of PDCD10 in the maintenance of homeostasis of a wide range of cells, the loss of gut epithelial PDCD10 may result in disruption of the colonic mucosal barrier, thus these individuals may be more aggressively affected by CCM diseases (54–57). In the somatic mutations, different genotypes or loci may also be associated with specific clinical phenotypes. For example, our preliminary results suggested that PIK3CA mutations may be associated with a higher risk of significant bleeding. Although the exact differences are not yet known, the revealing of the relationship between different mutation genotypes and clinical manifestations is an extremely interesting study, which may even guide the search for different therapeutic targets.
Strikingly, although advanced insights into the innovative knowledge of the CCM genes’ pathophysiological functions are emerging, the highly unpredictable clinical behaviors in individual patients still present a major challenge in clinical and basic research. Accumulated evidence clearly illustrated that, at least in a few cases, gene mutations alone are not sufficient to trigger CCM onset and progression (58–60). Moreover, it is also important that the highly variable severity of clinical symptoms is common, even among familial patients who carried the same genetic mutation. It has been found that in monozygotic twins who suffered the same germline mutation, exact similarity of the disease onset and clinical severity could be observed, in contrast to the different clinical severity of non-twin siblings in wide intrafamily (61). Notably, sometimes many suffered individuals could be clinically asymptomatic for a long time or even for their entire life. Once symptoms appear, there could be disastrous consequences, and the healthy quality of life could be seriously affected. Thus, what the initiator from the existence of lesions to the appearance of clinical symptoms is, and what causes the same initial encounter but different clinical outcomes, are interesting issues that we are trying to solve. The perspectives from the immune and inflammatory events described in the next part may point out a new direction for our thinking.
Immune and Inflammatory Mechanisms for CCM Formation
The CCM lesions should be regarded as the results of a great number of additional events. Indeed, inflammatory perturbations associated with the immune microenvironment might be one of the most fascinating concerns (62). Recent study results allowed the identification of major consequences and underlined the immune-related factors’ contribution to the CCM pathologic progression. In this part, we mainly followed these insights to draw the outline of the relationship between the CCMs and related immune and inflammatory events.
The Influence of Microbiome Is an Interesting Phenomenon
The microbiome has become a hot research topic in recent years, and has been identified to be associated with many diseases. However, the potential mechanisms through which it contributes to disease pathogenesis are still unclear. The works of Kahn et al. are particularly interesting and significant (45, 63). Firstly, the resistance to CCM formation was observed accidentally in mouse model of C57BL/6J background, which was previously identified as having 100% CCM penetrance. Then they demonstrated that, to stimulate the CCM formation, the bacterial microbiome is one of the primary sources. Meanwhile, they also revealed that, even the qualitative differences in the gut microbiome are small, they could have marked effects on the process of CCM development in animal models (45). Furthermore, the key role of TLR4in regulating inflammatory pathways in the immune microenvironment was highlighted. As their results suggested, the CCM formation would be driven by the activation of TLR4 receptors in endothelial cells, and the gut bacterial microbiome might be the leading actor (45). Given that the gut-brain axis might be a potential target, therapeutic exploration is being attempted. Obviously, the CCM formation could be inhibited in mice group which received pharmacological TLR4 blocking intervention. Similarly, the susceptibility could also be attenuated in germ-free mice, as well as in antibiotic-treated mice (63). In line with this, factors that affected the gut epithelial barrier homeostasis, involving chemical and genetic disruptions, and even colonic mucus alterations, can precipitate CCM development. As they revealed, genetic depletion of PDCD10 could impair gut barrier homeostasis, then accelerating the CCM formation (63). The interesting results of this study are fascinating, inspiring further studies to be continued (64, 65). In this light, it is reasonable to think that the effects of individual microbiomes may help to explain clinical variability. To test this hypothesis and determine whether the manipulation of gut microbiome-host interactions is a viable therapeutic strategy, future studies are urgently needed. In addition, it is necessary to reconsider whether strategies such as protection of gastrointestinal mucosal and prevention of gastrointestinal infection should be included in the clinical management of CCM patients.
Cumulative Direct Evidence Established the Role of Immunization
To identify what immune-related genetic modifiers would be associated with disease progression and severity, excellent research has been performed in recent decades. The Awad’s Team made an outstanding contribution to this field. Firstly, the exploration of the relationship between different gene expressions and clinical manifestations laid the basis for the gene spectrum of CCM research, which was also the origin of the discovery of immune-related molecules (52, 66, 67). Then the concepts and inflammation hypothesis in the pathogenesis of CCMs were first proposed, with laboratory investigation results (58, 68). By assessing the infiltrated immune cells and the immunoglobulin isotypes predominant in CCM lesions, they systematically described the immune responses in human CCMs. In addition, the evidence of co-localized IgG and complement membrane attack complexes in CCM lesions were reported (69). Then, the in-situ B-cell clonal expansion was identified, indicating antigenic stimulation in CCMs (69). Given that plasma cell-mediated immunity might be one contributory factor to CCM lesion, the evaluation of the effects of the immune response inhibition on the CCM lesions’ formation and maturation were done (70). Evidence suggested that B-cell depletion would have therapeutic benefits for CCM disease. Meanwhile, these findings also remind us that these biologics could be added into the list as potential therapeutic agents (70).
Recent results take this research to a new level (71). By using laser capture micro-dissection, plasma cells from CCM lesions were obtained, and mAbs (monoclonal antibody) were subsequently generated. Then antigens targeted by resident plasma cells in CCMs were characterized via detailed experiments and analysis (71). Despite the limitation of small sample size and the uncertainty of whether the mAbs would react with other antibodies, these data have given us a novel insight into the immunopathogenesis of CCM. In another piece of research, the infiltration of different subsets of leukocytes in CCM was revealed (72). Through RNA-seq analysis, increased inflammation-related genes were observed in the endothelial cells from CCM mouse model (72). Notably, the cytokines which perform the roles of inflammatory and immune cells’ recruitment, involving proinflammatory cytokines, chemokines, and adhesion molecules, are closely related to the differentially expressed genes. More importantly, neutrophils were found to have infiltrated into the CCM lesions, in the way of neutrophil extracellular traps formation (72). These findings were also validated in clinical specimens, which undoubtedly opened up new research ideas for researchers in this study field.
Advances in sequencing techniques and multi-omics analysis made it possible to access a more comprehensive understanding of the genetics and modification molecules of CCM. By using gene microarray technology, 78 abnormal expression genes related to inflammatory response in CCM lesions were observed, including 13 overexpressed immunoglobulin genes (66). Compared with control samples, probe sets revealed an up-regulation of 10 Ig genes and a distinct allele of the major histocompatibility complex in CCM samples, providing potential evidence for a characterized immune background in the CCM lesions (66). Furthermore, polymorphisms in genes of the immune-inflammatory response were also proven to be correlated with the severity of CCM disease (59, 73). Though further studies in larger sample sizes are needed, these results primarily suggested that immune-related factors might be potential predictors for phenotype variability and disease severity.
The identification of the accumulated immunologic cells could also contribute to the direct evidence. In a previous in vitro study, the infiltration of macrophage inflammatory cells in and around CCM lesions was reported, especially under the reaction of acute hemorrhage (74). In mouse models, macrophages were also proven in the CCM lesions by histological analysis (75). Similarly, comprehensive transcriptome analysis also provided strong support for the evidence of immune microenvironment in CCM pathology. Through the analysis of the immune microenvironment, it was demonstrated that nearly 30% of the total cell number were accounted by immune cells, and macrophages, NK cells, monocytes, as well as B cells and T cells were all included (76–78). Microglia, as resident immune cells of the central nervous system, have also attracted much attention (79). The microglia activation within the perilesional tissue were thought to mediate hematoma resolution (80, 81). Meanwhile, microglia are also thought to be involved in the pathological processes associated with epilepsy (82, 83).
The other thing that could also link CCM to the immune response is inflammatory susceptibility. Research in animal models have revealed that the anti-inflammatory threshold was decreased when the patient suffered CCM diseases (84). Thus, there is reason to believe that an enhanced inflammatory response could also be presented in CCM individuals. Or to put it another way, patients with CCMs have an increased susceptibility to inflammation. As low fluid shear stress could be one of the characteristics of CCM lesions, it is reasonable to consider whether these conditions could contribute to the increased susceptibility to inflammation and immune responses (85). Evidence from clinical blood samples is also suggestive. In a clinical study, it was suggested that inflammatory factors in plasma samples may be associated with clinical symptoms, such as epilepsy, a common hospital admitted complaint (86).
Taken together, all these results suggested an intrinsic immune response in CCMs. In addition, modifiers associated with genetic and epigenetic aspects determined inter-individual differences in susceptibility to stressful conditions. More detailed mechanisms should be further characterized, and potential therapeutic targets remain to be identified.
Immune and Inflammatory Mechanisms for Complications
Recurrent microbleeds, as we described previously, may be one of the most common complications of CCM (11, 12, 16, 87–90). Although the potential roles of the immune response in this disease have not been illustrated clearly, the inflammatory microenvironment following hemorrhagic events has been noted (20, 58, 91). The thrombosis and leaky blood-brain barrier would lead to the formation of a unique antigenic milieu for CCM lesions. On the one hand, it may regulate angiogenesis, leading to further progression of lesions. On the other hand, the risk of rebleeding would be further increased. Evidence from clinical samples and neuroimages defined the immune microenvironment in the characteristic vascular phenotype (58, 92, 93). Clearly, repeated hemorrhagic events could be partially regarded as the ‘trigger’ of the immune response. The defected blood brain barrier might be responsible for repeated leakages, and segregated caverns at various tissue stages were accumulated with chronic blood deposition and degradation products (94). The typical pathologic structures were filled with immune and inflammatory factors. Consequently, the prognosis, whether benign or malignant, is the result of immunological struggle.
The relationship between hemorrhagic events and inflammatory responses has been well described in multiple cerebrovascular anomalies, including aneurysm (95, 96), cerebral arteriovenous malformation (97), cerebral amyloid angiopathy (98, 99), and ischemic cerebrovascular disorders (100, 101). Notably, acute brain hemorrhages may not have an opportunity to lead a similar milieu with the longstanding deposition of blood products, because it would be absorbed over a normal length of time. The recurrent and chronic bleeding might be a unique characterization of the CCM lesion, and it is the long-term inflammatory cytokines that may cause further damage to the blood-brain barrier. Anti-inflammatory treatments seem to be working, by reducing bleed rate and severity. However, it is not easy to know whether the inflammation is prior to a hemorrhagic event or if the inflammatory response is resulted from bleeding. Interestingly, it is clear that innate inflammatory response could be presented with CCM, and inflammatory cells could also be found accumulating around the lesion. Thus, similar attempts of local immunologic manipulation could be encouraged to explore the novel therapeutic approaches for CCMs.
The individual who suffered CCMs was also predisposed to a lifetime risk of epilepsy. Even a single seizure may greatly decrease health-related life quality (102, 103). The hemosiderin deposition has been considered as one of the pathological backgrounds. The lesion size and location, as well as supratentorial mass effects, may be considered as risk factors. However, for whatever reason, the existence of the immune microenvironment is underpinned, and seizures are undoubtedly the result of the BBB impairment, molecule accumulation, and neuronal hyperexcitability (104). A very strong causal effect has been reported in BBB damage and ictal activities’ generating and sustaining. Distinct mechanisms, involving disequilibrium of ionic concentrations (105–109), accumulation of serum proteins (110–112), and metabolic mismatch of amino acids (113–115), have been confirmed to induce neuronal hyperexcitability during BBB dysfunction. In turn, seizures may further exacerbate this vicious cycle of pathological cascades. For example, given ISF (interstitial fluid) formation and movement would be interfered with by damaged BBB during seizures, the expulsion of waste substances that affect neuronal electrophysiological activity would be impacted (116–118). Therefore, it is plausible to assume that the characteristic immune microenvironment of CCM may be the pathological background of epileptic seizures, and repeated or persistent seizures can worsen this milieu.
Some studies are even ahead of their time. Evidence from inflammatory plasma biomarkers, which reflected the seizures and hemorrhagic activities, were gradually reported (86, 119). These results predictively implied the association between inflammatory cytokines and subsequent clinical activities. Meanwhile, several drugs were clinically tested via a multivariate network (120–122). Although there are still hurdles to overcome, these preclinical discoveries and biomarker validations may contribute to future therapeutic targets’ exploration.
Prospects and Hypothesis for Future Research
Benefited from the convergence of multiple scientific and medical advances, so much progress has been made in the CCM research field in the past two decades. Germ line mutations and/or somatic mutations are the causative factors, which played major roles in endothelial homeostasis disequilibrium events. However, the whole pathogenesis is a continuum involving lesion development and growth. On the macro level, our bodies are constantly exposed to an antigenic environment. On the micro, CCM lesions also exist in the immune microenvironment (Figure 1). Numerous factors involving the microbiome could affect the gut-brain axis immune system, which would affect the overall immune state of the body, diversifying the severity of the disease. Meanwhile, polymorphic gene hubs, especially about the inflammation-related genes, contributed to CCM development and progression during the distinct phase. For CCM lesions, acute and chronic bleeding are both the results and the beginnings of a new vicious cycle in the context of the immune microenvironment. Moreover, the CCM pathological context itself is an immune response site. All the molecular activities at the micro-immunological levels are closely related to clinical outcomes.
To sum up, our work emphasized the importance of immune factors as genetic modifiers, providing insights into the search for new potential therapeutic targets. Strategies preventing detrimental immune responses and enhancing beneficial immune responses are being tested, and these efforts are highly expected to make targeted and efficient clinical transformation.
Author Contributions
TT and ZP contributed equally to this work and should be considered to be co-first authors. TT reviewed related literatures and drafted the paper. ZP reviewed related literatures and drew the illustration figures. JR discussed important points and made important revisions to the paper. HZ reviewed the final version and approved the paper for publication. All authors contributed to the article and approved the submitted version.
Funding
National Natural Science Foundation of China Award Number: 81971113
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Thanks to the research teams who have made outstanding contributions to the CCM research field.
References
1. Hong T, Xiao X, Ren J, Cui B, Zong Y, Zou J, et al. Somatic MAP3K3 and PIK3CA Mutations in Sporadic Cerebral and Spinal Cord Cavernous Malformations. Brain (2021) 144:2648–58. doi: 10.1093/brain/awab117
2. Paddock M, Lanham S, Gill K, Sinha S, Connolly DJA. Pediatric Cerebral Cavernous Malformations. Pediatr Neurol (2021) 116:74–83. doi: 10.1016/j.pediatrneurol.2020.11.004
3. Zafar A, Quadri SA, Farooqui M, Ikram A, Robinson M, Hart BL, et al. Familial Cerebral Cavernous Malformations. Stroke (2019) 50:1294–301. doi: 10.1161/STROKEAHA.118.022314
4. Ren AA, Snellings DA, Su YS, Hong CC, Castro M, Tang AT, et al. PIK3CA and CCM Mutations Fuel Cavernomas Through a Cancer-Like Mechanism. Nature (2021) 594:271–6. doi: 10.1038/s41586-021-03562-8
5. Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, et al. EndMT Contributes to the Onset and Progression of Cerebral Cavernous Malformations. Nature (2013) 498:492–6. doi: 10.1038/nature12207
6. Zhou Z, Tang AT, Wong WY, Bamezai S, Goddard LM, Shenkar R, et al. Cerebral Cavernous Malformations Arise From Endothelial Gain of MEKK3-KLF2/4 Signalling. Nature (2016) 532:122–6. doi: 10.1038/nature17178
7. Al-Shahi Salman R, Hall JM, Horne MA, Moultrie F, Josephson CB, Bhattacharya JJ, et al. Untreated Clinical Course of Cerebral Cavernous Malformations: A Prospective, Population-Based Cohort Study. Lancet Neurol (2012) 11:217–24. doi: 10.1016/S1474-4422(12)70004-2
8. Flemming KD. Predicting the Clinical Behaviour of Cavernous Malformations. Lancet Neurol (2012) 11:202–3. doi: 10.1016/S1474-4422(12)70016-9
9. Horne MA, Flemming KD, Su IC, Stapf C, Jeon JP, Li D, et al. Clinical Course of Untreated Cerebral Cavernous Malformations: A Meta-Analysis of Individual Patient Data. Lancet Neurol (2016) 15:166–73. doi: 10.1016/S1474-4422(15)00303-8
10. Rigamonti D, Hadley MN, Drayer BP, Johnson PC, Hoenig-Rigamonti K, Knight JT, et al. Cerebral Cavernous Malformations. Incidence and Familial Occurrence. N Engl J Med (1988) 319:343–7. doi: 10.1056/NEJM198808113190605
11. Al-Shahi Salman R, Berg MJ, Morrison L, Awad IA, Angioma Alliance Scientific Advisory B. Hemorrhage From Cavernous Malformations of the Brain: Definition and Reporting Standards. Angioma Alliance Scientific Advisory Board. Stroke (2008) 39:3222–30. doi: 10.1161/STROKEAHA.108.515544
12. Flemming KD, Link MJ, Christianson TJ, Brown RD Jr. Prospective Hemorrhage Risk of Intracerebral Cavernous Malformations. Neurology (2012) 78:632–6. doi: 10.1212/WNL.0b013e318248de9b
13. Joseph NK, Kumar S, Brown RD Jr., Lanzino G, Flemming KD. Influence of Pregnancy on Hemorrhage Risk in Women With Cerebral and Spinal Cavernous Malformations. Stroke (2021) 52:434–41. doi: 10.1161/STROKEAHA.120.031761
14. Li DY, Whitehead KJ. Evaluating Strategies for the Treatment of Cerebral Cavernous Malformations. Stroke (2010) 41:S92–4. doi: 10.1161/STROKEAHA.110.594929
15. Li D, Wu ZY, Liu PP, Ma JP, Huo XL, Wang L, et al. Natural History of Brainstem Cavernous Malformations: Prospective Hemorrhage Rate and Adverse Factors in a Consecutive Prospective Cohort. J Neurosurg (2020) 134:917–28. doi: 10.3171/2019.12.JNS192856
16. Kashefiolasl S, Bruder M, Brawanski N, Herrmann E, Seifert V, Tritt S, et al. A Benchmark Approach to Hemorrhage Risk Management of Cavernous Malformations. Neurology (2018) 90:e856–63. doi: 10.1212/WNL.0000000000005066
17. Flemming KD, Lanzino G. Cerebral Cavernous Malformation: What a Practicing Clinician Should Know. Mayo Clin Proc (2020) 95:2005–20. doi: 10.1016/j.mayocp.2019.11.005
18. Jehi LE, Palmini A, Aryal U, Coras R, Paglioli E. Cerebral Cavernous Malformations in the Setting of Focal Epilepsies: Pathological Findings, Clinical Characteristics, and Surgical Treatment Principles. Acta Neuropathol (2014) 128:55–65. doi: 10.1007/s00401-014-1294-y
19. Snellings DA, Hong CC, Ren AA, Lopez-Ramirez MA, Girard R, Srinath A, et al. Cerebral Cavernous Malformation: From Mechanism to Therapy. Circ Res (2021) 129:195–215. doi: 10.1161/CIRCRESAHA.121.318174
20. Rustenhoven J, Tanumihardja C, Kipnis J. Cerebrovascular Anomalies: Perspectives From Immunology and Cerebrospinal Fluid Flow. Circ Res (2021) 129:174–94. doi: 10.1161/CIRCRESAHA.121.318173
21. Weng J, Yang Y, Song D, Huo R, Li H, Chen Y, et al. Somatic MAP3K3 Mutation Defines a Subclass of Cerebral Cavernous Malformation. Am J Hum Genet (2021) 108:942–50. doi: 10.1016/j.ajhg.2021.04.005
22. Retta SF, Perrelli A, Trabalzini L, Finetti F. From Genes and Mechanisms to Molecular-Targeted Therapies: The Long Climb to the Cure of Cerebral Cavernous Malformation (CCM) Disease. Methods Mol Biol (2020) 2152:3–25. doi: 10.1007/978-1-0716-0640-7_1
23. Lucas M, Costa AF, Montori M, Solano F, Zayas MD, Izquierdo G. Germline Mutations in the CCM1 Gene, Encoding Krit1, Cause Cerebral Cavernous Malformations. Ann Neurol (2001) 49:529–32. doi: 10.1002/ana.105
24. Zawistowski JS, Serebriiskii IG, Lee MF, Golemis EA, Marchuk DA. KRIT1 Association With the Integrin-Binding Protein ICAP-1: A New Direction in the Elucidation of Cerebral Cavernous Malformations (CCM1) Pathogenesis. Hum Mol Genet (2002) 11:389–96. doi: 10.1093/hmg/11.4.389
25. Sahoo T, Johnson EW, Thomas JW, Kuehl PM, Jones TL, Dokken CG, et al. Mutations in the Gene Encoding KRIT1, a Krev-1/Rap1a Binding Protein, Cause Cerebral Cavernous Malformations (CCM1). Hum Mol Genet (1999) 8:2325–33. doi: 10.1093/hmg/8.12.2325
26. Liu W, Draheim KM, Zhang R, Calderwood DA, Boggon TJ. Mechanism for KRIT1 Release of ICAP1-Mediated Suppression of Integrin Activation. Mol Cell (2013) 49:719–29. doi: 10.1016/j.molcel.2012.12.005
27. Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, et al. Truncating Mutations in CCM1, Encoding KRIT1, Cause Hereditary Cavernous Angiomas. Nat Genet (1999) 23:189–93. doi: 10.1038/13815
28. Denier C, Goutagny S, Labauge P, Krivosic V, Arnoult M, Cousin A, et al. Mutations Within the MGC4607 Gene Cause Cerebral Cavernous Malformations. Am J Hum Genet (2004) 74:326–37. doi: 10.1086/381718
29. Liquori CL, Berg MJ, Squitieri F, Leedom TP, Ptacek L, Johnson EW, et al. Deletions in CCM2 are a Common Cause of Cerebral Cavernous Malformations. Am J Hum Genet (2007) 80:69–75. doi: 10.1086/510439
30. Boulday G, Rudini N, Maddaluno L, Blecon A, Arnould M, Gaudric A, et al. Developmental Timing of CCM2 Loss Influences Cerebral Cavernous Malformations in Mice. J Exp Med (2011) 208:1835–47. doi: 10.1084/jem.20110571
31. Harel L, Costa B, Tcherpakov M, Zapatka M, Oberthuer A, Hansford LM, et al. CCM2 Mediates Death Signaling by the TrkA Receptor Tyrosine Kinase. Neuron (2009) 63:585–91. doi: 10.1016/j.neuron.2009.08.020
32. Cullere X, Plovie E, Bennett PM, MacRae CA, Mayadas TN. The Cerebral Cavernous Malformation Proteins CCM2L and CCM2 Prevent the Activation of the MAP Kinase MEKK3. Proc Natl Acad Sci USA (2015) 112:14284–9. doi: 10.1073/pnas.1510495112
33. Vannier DR, Shapeti A, Chuffart F, Planus E, Manet S, Rivier P, et al. CCM2-Deficient Endothelial Cells Undergo a ROCK-Dependent Reprogramming Into Senescence-Associated Secretory Phenotype. Angiogenesis (2021) 24:843–60. doi: 10.1007/s10456-021-09809-2
34. Zheng X, Xu C, Di Lorenzo A, Kleaveland B, Zou Z, Seiler C, et al. CCM3 Signaling Through Sterile 20-Like Kinases Plays an Essential Role During Zebrafish Cardiovascular Development and Cerebral Cavernous Malformations. J Clin Invest (2010) 120:2795–804. doi: 10.1172/JCI39679
35. Jenny Zhou H, Qin L, Zhang H, Tang W, Ji W, He Y, et al. Endothelial Exocytosis of Angiopoietin-2 Resulting From CCM3 Deficiency Contributes to Cerebral Cavernous Malformation. Nat Med (2016) 22:1033–42. doi: 10.1038/nm.4169
36. Zhou HJ, Qin L, Jiang Q, Murray KN, Zhang H, Li B, et al. Caveolae-Mediated Tie2 Signaling Contributes to CCM Pathogenesis in a Brain Endothelial Cell-Specific Pdcd10-Deficient Mouse Model. Nat Commun (2021) 12:504. doi: 10.1038/s41467-020-20774-0
37. Peyre M, Miyagishima D, Bielle F, Chapon F, Sierant M, Venot Q, et al. Somatic PIK3CA Mutations in Sporadic Cerebral Cavernous Malformations. N Engl J Med (2021) 385:996. doi: 10.1056/NEJMoa2100440
38. Venugopal V, Sumi S. Molecular Biomarkers and Drug Targets in Brain Arteriovenous and Cavernous Malformations: Where Are We? Stroke (2022) 53:279–89. doi: 10.1161/STROKEAHA.121.035654
39. Castro M, Lavina B, Ando K, Alvarez-Aznar A, Abu Taha A, Brakebusch C, et al. CDC42 Deletion Elicits Cerebral Vascular Malformations via Increased MEKK3-Dependent KLF4 Expression. Circ Res (2019) 124:1240–52. doi: 10.1161/CIRCRESAHA.118.314300
40. Fisher OS, Deng H, Liu D, Zhang Y, Wei R, Deng Y, et al. Structure and Vascular Function of MEKK3-Cerebral Cavernous Malformations 2 Complex. Nat Commun (2015) 6:7937. doi: 10.1038/ncomms8937
41. Zhou Z, Rawnsley DR, Goddard LM, Pan W, Cao XJ, Jakus Z, et al. The Cerebral Cavernous Malformation Pathway Controls Cardiac Development via Regulation of Endocardial MEKK3 Signaling and KLF Expression. Dev Cell (2015) 32:168–80. doi: 10.1016/j.devcel.2014.12.009
42. Castillo SD, Baselga E, Graupera M. PIK3CA Mutations in Vascular Malformations. Curr Opin Hematol (2019) 26:170–8. doi: 10.1097/MOH.0000000000000496
43. Rodriguez-Laguna L, Agra N, Ibanez K, Oliva-Molina G, Gordo G, Khurana N, et al. Somatic Activating Mutations in PIK3CA Cause Generalized Lymphatic Anomaly. J Exp Med (2019) 216:407–18. doi: 10.1084/jem.20181353
44. di Blasio L, Puliafito A, Gagliardi PA, Comunanza V, Somale D, Chiaverina G, et al. PI3K/mTOR Inhibition Promotes the Regression of Experimental Vascular Malformations Driven by PIK3CA-Activating Mutations. Cell Death Dis (2018) 9:45. doi: 10.1038/s41419-017-0064-x
45. Tang AT, Choi JP, Kotzin JJ, Yang Y, Hong CC, Hobson N, et al. Endothelial TLR4 and the Microbiome Drive Cerebral Cavernous Malformations. Nature (2017) 545:305–10. doi: 10.1038/nature22075
46. Polster SP, Sharma A, Tanes C, Tang AT, Mericko P, Cao Y, et al. Permissive Microbiome Characterizes Human Subjects With a Neurovascular Disease Cavernous Angioma. Nat Commun (2020) 11:2659. doi: 10.1038/s41467-020-16436-w
47. Brownlee WJ, Roxburgh R. Mystery Case: Cutaneous Lesions in KRIT1-Associated Cerebral Cavernous Malformations. Neurology (2014) 83:e133–4. doi: 10.1212/WNL.0000000000000849
48. Haghighi A, Fathi D, Shahbazi M, Motahari MM, Friedman B. Identification of a C.601C>G Mutation in the CCM1 Gene in a Kindred With Multiple Skin, Spinal and Cerebral Cavernous Malformations. J Neurol Sci (2013) 334:97–101. doi: 10.1016/j.jns.2013.07.2518
49. Musunuru K, Hillard VH, Murali R. Widespread Central Nervous System Cavernous Malformations Associated With Cafe-Au-Lait Skin Lesions. Case Report. J Neurosurg (2003) 99:412–5. doi: 10.3171/jns.2003.99.2.0412
50. Sirvente J, Enjolras O, Wassef M, Tournier-Lasserve E, Labauge P. Frequency and Phenotypes of Cutaneous Vascular Malformations in a Consecutive Series of 417 Patients With Familial Cerebral Cavernous Malformations. J Eur Acad Dermatol Venereol (2009) 23:1066–72. doi: 10.1111/j.1468-3083.2009.03263.x
51. Gianfrancesco F, Cannella M, Martino T, Maglione V, Esposito T, Innocenzi G, et al. Highly Variable Penetrance in Subjects Affected With Cavernous Cerebral Angiomas (CCM) Carrying Novel CCM1 and CCM2 Mutations. Am J Med Genet B Neuropsychiatr Genet (2007) 144B:691–5. doi: 10.1002/ajmg.b.30381
52. Gault J, Sain S, Hu LJ, Awad IA. Spectrum of Genotype and Clinical Manifestations in Cerebral Cavernous Malformations. Neurosurgery (2006) 59:1278–84. doi: 10.1227/01.NEU.0000249188.38409.03
53. Denier C, Labauge P, Bergametti F, Marchelli F, Riant F, Arnoult M, et al. Genotype-Phenotype Correlations in Cerebral Cavernous Malformations Patients. Ann Neurol (2006) 60:550–6. doi: 10.1002/ana.20947
54. Riant F, Bergametti F, Fournier HD, Chapon F, Michalak-Provost S, Cecillon M, et al. CCM3 Mutations Are Associated With Early-Onset Cerebral Hemorrhage and Multiple Meningiomas. Mol Syndromol (2013) 4:165–72. doi: 10.1159/000350042
55. Shenkar R, Shi C, Rebeiz T, Stockton RA, McDonald DA, Mikati AG, et al. Exceptional Aggressiveness of Cerebral Cavernous Malformation Disease Associated With PDCD10 Mutations. Genet Med (2015) 17:188–96. doi: 10.1038/gim.2014.97
56. Nardella G, Visci G, Guarnieri V, Castellana S, Biagini T, Bisceglia L, et al. A Single-Center Study on 140 Patients With Cerebral Cavernous Malformations: 28 New Pathogenic Variants and Functional Characterization of a PDCD10 Large Deletion. Hum Mutat (2018) 39:1885–900. doi: 10.1002/humu.23629
57. Valentino M, Dejana E, Malinverno M. The Multifaceted PDCD10/CCM3 Gene. Genes Dis (2021) 8:798–813. doi: 10.1016/j.gendis.2020.12.008
58. Shenkar R, Shi C, Check IJ, Lipton HL, Awad IA. Concepts and Hypotheses: Inflammatory Hypothesis in the Pathogenesis of Cerebral Cavernous Malformations. Neurosurgery (2007) 61:693–702; discussion 702-3. doi: 10.1227/01.NEU.0000298897.38979.07
59. Choquet H, Kim H. Genome-Wide Genotyping of Cerebral Cavernous Malformation Type 1 Individuals to Identify Genetic Modifiers of Disease Severity. Methods Mol Biol (2020) 2152:77–84. doi: 10.1007/978-1-0716-0640-7_6
60. Perrelli A, Retta SF. Polymorphisms in Genes Related to Oxidative Stress and Inflammation: Emerging Links With the Pathogenesis and Severity of Cerebral Cavernous Malformation Disease. Free Radic Biol Med (2021) 172:403–17. doi: 10.1016/j.freeradbiomed.2021.06.021
61. Dammann P, Hehr U, Weidensee S, Zhu Y, Gerlach R, Sure U. Two-Hit Mechanism in Cerebral Cavernous Malformation? A Case of Monozygotic Twins With a CCM1/KRIT1 Germline Mutation. Neurosurg Rev (2013) 36:483–6. doi: 10.1007/s10143-013-0456-z
62. Retta SF, Glading AJ. Oxidative Stress and Inflammation in Cerebral Cavernous Malformation Disease Pathogenesis: Two Sides of the Same Coin. Int J Biochem Cell Biol (2016) 81:254–70. doi: 10.1016/j.biocel.2016.09.011
63. Tang AT, Sullivan KR, Hong CC, Goddard LM, Mahadevan A, Ren A, et al. Distinct Cellular Roles for PDCD10 Define a Gut-Brain Axis in Cerebral Cavernous Malformation. Sci Transl Med 11 (2019) 11:eaaw3521. doi: 10.1126/scitranslmed.aaw3521
64. Abdelilah-Seyfried S, Tournier-Lasserve E, Derry WB. Blocking Signalopathic Events to Treat Cerebral Cavernous Malformations. Trends Mol Med (2020) 26:874–87. doi: 10.1016/j.molmed.2020.03.003
65. Starke RM, McCarthy DJ, Komotar RJ, Connolly ES. Gut Microbiome and Endothelial TLR4 Activation Provoke Cerebral Cavernous Malformations. Neurosurgery (2017) 81:N44–6. doi: 10.1093/neuros/nyx450
66. Shenkar R, Elliott JP, Diener K, Gault J, Hu LJ, Cohrs RJ, et al. Differential Gene Expression in Human Cerebrovascular Malformations. Neurosurgery (2003) 52:465–77; discussion 477-8. doi: 10.1227/01.neu.0000044131.03495.22
67. Gault J, Sarin H, Awadallah NA, Shenkar R, Awad IA. Pathobiology of Human Cerebrovascular Malformations: Basic Mechanisms and Clinical Relevance. Neurosurgery (2004) 55:1–16; discussion 16-7. doi: 10.1227/01.neu.0000440729.59133.c9
68. Shi C, Shenkar R, Batjer HH, Check IJ, Awad IA. Oligoclonal Immune Response in Cerebral Cavernous Malformations. Laboratory Investigation. J Neurosurg (2007) 107:1023–6. doi: 10.3171/JNS-07/11/1023
69. Shi C, Shenkar R, Kinloch A, Henderson SG, Shaaya M, Chong AS, et al. Immune Complex Formation and in Situ B-Cell Clonal Expansion in Human Cerebral Cavernous Malformations. J Neuroimmunol (2014) 272:67–75. doi: 10.1016/j.jneuroim.2014.04.016
70. Shi C, Shenkar R, Zeineddine HA, Girard R, Fam MD, Austin C, et al. B-Cell Depletion Reduces the Maturation of Cerebral Cavernous Malformations in Murine Models. J Neuroimmune Pharmacol (2016) 11:369–77. doi: 10.1007/s11481-016-9670-0
71. Zhang D, Kinloch AJ, Srinath A, Shenkar R, Girard R, Lightle R, et al. Antibodies in Cerebral Cavernous Malformations React With Cytoskeleton Autoantigens in the Lesional Milieu. J Autoimmun (2020) 113:102469. doi: 10.1016/j.jaut.2020.102469
72. Yau ACY, Globisch MA, Onyeogaziri FC, Conze LL, Smith R, Jauhiainen S, et al. Inflammation and Neutrophil Extracellular Traps in Cerebral Cavernous Malformation. Cell Mol Life Sci (2022) 79:206. doi: 10.1007/s00018-022-04224-2
73. Choquet H, Pawlikowska L, Nelson J, McCulloch CE, Akers A, Baca B, et al. Polymorphisms in Inflammatory and Immune Response Genes Associated With Cerebral Cavernous Malformation Type 1 Severity. Cerebrovasc Dis (2014) 38:433–40. doi: 10.1159/000369200
74. Vymazal J, Brooks RA, Baumgarner C, Tran V, Katz D, Bulte JW, et al. The Relation Between Brain Iron and NMR Relaxation Times: An In Vitro Study. Magn Reson Med (1996) 35:56–61. doi: 10.1002/mrm.1910350108
75. Plummer NW, Gallione CJ, Srinivasan S, Zawistowski JS, Louis DN, Marchuk DA. Loss of P53 Sensitizes Mice With a Mutation in Ccm1 (KRIT1) to Development of Cerebral Vascular Malformations. Am J Pathol (2004) 165:1509–18. doi: 10.1016/S0002-9440(10)63409-8
76. Li Y, Yang J, Huang Y, Ge S, Song X, Jia R, et al. Cellular Heterogeneity and Immune Microenvironment Revealed by Single-Cell Transcriptome in Venous Malformation and Cavernous Venous Malformation. J Mol Cell Cardiol (2022) 162:130–43. doi: 10.1016/j.yjmcc.2021.09.004
77. Koskimaki J, Zhang D, Li Y, Saadat L, Moore T, Lightle R, et al. Transcriptome Clarifies Mechanisms of Lesion Genesis Versus Progression in Models of Ccm3 Cerebral Cavernous Malformations. Acta Neuropathol Commun (2019) 7:132. doi: 10.1186/s40478-019-0789-0
78. Koskimaki J, Girard R, Li Y, Saadat L, Zeineddine HA, Lightle R, et al. Comprehensive Transcriptome Analysis of Cerebral Cavernous Malformation Across Multiple Species and Genotypes. JCI Insight (2019) 4:e126167. doi: 10.1172/jci.insight.126167
79. Timis TL, Florian IA, Susman S, Florian IS. Involvement of Microglia in the Pathophysiology of Intracranial Aneurysms and Vascular Malformations-A Short Overview. Int J Mol Sci (2021) 22:6141. doi: 10.3390/ijms22116141
80. Lyu J, Xie D, Bhatia TN, Leak RK, Hu X, Jiang X. Microglial/Macrophage Polarization and Function in Brain Injury and Repair After Stroke. CNS Neurosci Ther (2021) 27:515–27. doi: 10.1111/cns.13620
81. Zhao X, Grotta J, Gonzales N, Aronowski J. Hematoma Resolution as a Therapeutic Target: The Role of Microglia/Macrophages. Stroke (2009) 40:S92–4. doi: 10.1161/STROKEAHA.108.533158
82. Boer K, Spliet WG, van Rijen PC, Redeker S, Troost D, Aronica E. Evidence of Activated Microglia in Focal Cortical Dysplasia. J Neuroimmunol (2006) 173:188–95. doi: 10.1016/j.jneuroim.2006.01.002
83. Liu J, Reeves C, Michalak Z, Coppola A, Diehl B, Sisodiya SM, et al. Evidence for mTOR Pathway Activation in a Spectrum of Epilepsy-Associated Pathologies. Acta Neuropathol Commun (2014) 2:71. doi: 10.1186/2051-5960-2-71
84. Corr M, Lerman I, Keubel JM, Ronacher L, Misra R, Lund F, et al. Decreased Krev Interaction-Trapped 1 Expression Leads to Increased Vascular Permeability and Modifies Inflammatory Responses In Vivo. Arterioscler Thromb Vasc Biol (2012) 32:2702–10. doi: 10.1161/ATVBAHA.112.300115
85. Li J, Zhao Y, Coleman P, Chen J, Ting KK, Choi JP, et al. Low Fluid Shear Stress Conditions Contribute to Activation of Cerebral Cavernous Malformation Signalling Pathways. Biochim Biophys Acta Mol Basis Dis (2019) 1865:165519. doi: 10.1016/j.bbadis.2019.07.013
86. Girard R, Zeineddine HA, Fam MD, Mayampurath A, Cao Y, Shi C, et al. Plasma Biomarkers of Inflammation Reflect Seizures and Hemorrhagic Activity of Cerebral Cavernous Malformations. Transl Stroke Res (2018) 9:34–43. doi: 10.1007/s12975-017-0561-3
87. Moultrie F, Horne MA, Josephson CB, Hall JM, Counsell CE, Bhattacharya JJ, et al. Outcome After Surgical or Conservative Management of Cerebral Cavernous Malformations. Neurology (2014) 83:582–9. doi: 10.1212/WNL.0000000000000684
88. Gross BA, Du R. Hemorrhage From Cerebral Cavernous Malformations: A Systematic Pooled Analysis. J Neurosurg (2017) 126:1079–87. doi: 10.3171/2016.3.JNS152419
89. Gross BA, Jankowitz BT, Friedlander RM. Cerebral Intraparenchymal Hemorrhage: A Review. JAMA (2019) 321:1295–303. doi: 10.1001/JAMA.2019.2413
90. Chen B, Herten A, Saban D, Rauscher S, Radbruch A, Schmidt B, et al. Hemorrhage From Cerebral Cavernous Malformations: The Role of Associated Developmental Venous Anomalies. Neurology (2020) 95:e89–96. doi: 10.1212/WNL.0000000000009730
91. Perrelli A, Fatehbasharzad P, Benedetti V, Ferraris C, Fontanella M, De Luca E, et al. Towards Precision Nanomedicine for Cerebrovascular Diseases With Emphasis on Cerebral Cavernous Malformation (CCM). Expert Opin Drug Delivery (2021) 18:849–76. doi: 10.1080/17425247.2021.1873273
92. Robinson JR Jr., Awad IA, Masaryk TJ, Estes ML. Pathological Heterogeneity of Angiographically Occult Vascular Malformations of the Brain. Neurosurgery (1993) 33:547–54; discussion 554-5. doi: 10.1227/00006123-199310000-00001
93. Rigamonti D, Drayer BP, Johnson PC, Hadley MN, Zabramski J, Spetzler RF. The MRI Appearance of Cavernous Malformations (Angiomas). J Neurosurg (1987) 67:518–24. doi: 10.3171/jns.1987.67.4.0518
94. Steiger HJ, Markwalder TM, Reulen HJ. Clinicopathological Relations of Cerebral Cavernous Angiomas: Observations in Eleven Cases. Neurosurgery (1987) 21:879–84. doi: 10.1227/00006123-198712000-00016
95. Bevers MB, Wolcott Z, Bache S, Hansen C, Sastre C, Mylvaganam R, et al. Soluble ST2 Links Inflammation to Outcome After Subarachnoid Hemorrhage. Ann Neurol (2019) 86:384–94. doi: 10.1002/ana.25545
96. Badjatia N, Monahan A, Carpenter A, Zimmerman J, Schmidt JM, Claassen J, et al. Inflammation, Negative Nitrogen Balance, and Outcome After Aneurysmal Subarachnoid Hemorrhage. Neurology (2015) 84:680–7. doi: 10.1212/WNL.0000000000001259
97. Winkler EA, Kim CN, Ross JM, Garcia JH, Gil E, Oh I, et al. A Single-Cell Atlas of the Normal and Malformed Human Brain Vasculature. Science (2022) 375:eabi7377. doi: 10.1126/science.abi7377
98. Aranha MR, Fortea J, Carmona-Iragui M. Cerebral Amyloid Angiopathy-Related Inflammation in Down Syndrome-Related Alzheimer Disease. Neurology (2022) 98: 1021–2. doi: 10.1212/WNL.0000000000200704
99. Auriel E, Charidimou A, Gurol ME, Ni J, Van Etten ES, Martinez-Ramirez S, et al. Validation of Clinicoradiological Criteria for the Diagnosis of Cerebral Amyloid Angiopathy-Related Inflammation. JAMA Neurol (2016) 73:197–202. doi: 10.1001/jamaneurol.2015.4078
100. Shi K, Tian DC, Li ZG, Ducruet AF, Lawton MT, Shi FD. Global Brain Inflammation in Stroke. Lancet Neurol (2019) 18:1058–66. doi: 10.1016/S1474-4422(19)30078-X
101. Flemming A. Calming Inflammation to Prevent Stroke Damage. Nat Rev Immunol (2019) 19:473. doi: 10.1038/s41577-019-0197-5
102. Josephson CB, Leach JP, Duncan R, Roberts RC, Counsell CE, Al-Shahi Salman R, et al. Seizure Risk From Cavernous or Arteriovenous Malformations: Prospective Population-Based Study. Neurology (2011) 76:1548–54. doi: 10.1212/WNL.0b013e3182190f37
103. Fox CK, Nelson J, McCulloch CE, Weinsheimer S, Pawlikowska L, Hart B, et al. Seizure Incidence Rates in Children and Adults With Familial Cerebral Cavernous Malformations. Neurology (2021) 97:e1210-6. doi: 10.1212/WNL.0000000000012569
104. Noe FM, Marchi N. Central Nervous System Lymphatic Unit, Immunity, and Epilepsy: Is There a Link? Epilepsia Open (2019) 4:30–9. doi: 10.1002/epi4.12302
105. Weiss JH, Sensi SL, Koh JY. Zn(2+): A Novel Ionic Mediator of Neural Injury in Brain Disease. Trends Pharmacol Sci (2000) 21:395–401. doi: 10.1016/s0165-6147(00)01541-8
106. Proix T, Bartolomei F, Chauvel P, Bernard C, Jirsa VK. Permittivity Coupling Across Brain Regions Determines Seizure Recruitment in Partial Epilepsy. J Neurosci (2014) 34:15009–21. doi: 10.1523/JNEUROSCI.1570-14.2014
107. Buchin A, Chizhov A, Huberfeld G, Miles R, Gutkin BS. Reduced Efficacy of the KCC2 Cotransporter Promotes Epileptic Oscillations in a Subiculum Network Model. J Neurosci (2016) 36:11619–33. doi: 10.1523/JNEUROSCI.4228-15.2016
108. Gentiletti D, Suffczynski P, Gnatkovsky V, de Curtis M. Changes of Ionic Concentrations During Seizure Transitions - A Modeling Study. Int J Neural Syst (2017) 27:1750004. doi: 10.1142/S0129065717500046
109. Ridley B, Marchi A, Wirsich J, Soulier E, Confort-Gouny S, Schad L, et al. Brain Sodium MRI in Human Epilepsy: Disturbances of Ionic Homeostasis Reflect the Organization of Pathological Regions. Neuroimage (2017) 157:173–83. doi: 10.1016/j.neuroimage.2017.06.011
110. Kazamatsuri H. Elevated Serum Alkaline Phosphatase Levels in Epilepsy During Diphenylhydantoin Therapy. N Engl J Med (1970) 283:1411–2. doi: 10.1056/NEJM197012172832518
111. Kodama Y, Kodama H, Kuranari M, Tsutsumi K, Ono S, Fujimura A. No Effect of Gender or Age on Binding Characteristics of Valproic Acid to Serum Proteins in Pediatric Patients With Epilepsy. J Clin Pharmacol (1999) 39:1070–6. doi: 10.1177/00912709922011728
112. Lin Z, Gu Y, Zhou R, Wang M, Guo Y, Chen Y, et al. Serum Exosomal Proteins F9 and TSP-1 as Potential Diagnostic Biomarkers for Newly Diagnosed Epilepsy. Front Neurosci (2020) 14:737. doi: 10.3389/fnins.2020.00737
113. Seal RP, Amara SG. Excitatory Amino Acid Transporters: A Family in Flux. Annu Rev Pharmacol Toxicol (1999) 39:431–56. doi: 10.1146/annurev.pharmtox.39.1.431
114. Olson CA, Vuong HE, Yano JM, Liang QY, Nusbaum DJ, Hsiao EY. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell (2018) 173:1728–41.e13. doi: 10.1016/j.cell.2018.04.027
115. Marafi D, Fatih JM, Kaiyrzhanov R, Ferla MP, Gijavanekar C, Al-Maraghi A, et al. Biallelic Variants in SLC38A3 Encoding a Glutamine Transporter Cause Epileptic Encephalopathy. Brain (2021) 145:909–24. doi: 10.1093/brain/awab369
116. Marchi N, Banjara M, Janigro D. Blood-Brain Barrier, Bulk Flow, and Interstitial Clearance in Epilepsy. J Neurosci Methods (2016) 260:118–24. doi: 10.1016/j.jneumeth.2015.06.011
117. Lange C, Storkebaum E, de Almodovar CR, Dewerchin M, Carmeliet P. Vascular Endothelial Growth Factor: A Neurovascular Target in Neurological Diseases. Nat Rev Neurol (2016) 12:439–54. doi: 10.1038/nrneurol.2016.88
118. Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol Rev (2019) 99:21–78. doi: 10.1152/physrev.00050.2017
119. Girard R, Zeineddine HA, Koskimaki J, Fam MD, Cao Y, Shi C, et al. Plasma Biomarkers of Inflammation and Angiogenesis Predict Cerebral Cavernous Malformation Symptomatic Hemorrhage or Lesional Growth. Circ Res (2018) 122:1716–21. doi: 10.1161/CIRCRESAHA.118.312680
120. Polster SP, Cao Y, Carroll T, Flemming K, Girard R, Hanley D, et al. Trial Readiness in Cavernous Angiomas With Symptomatic Hemorrhage (CASH). Neurosurgery (2019) 84:954–64. doi: 10.1093/neuros/nyy108
121. Girard R, Li Y, Stadnik A, Shenkar R, Hobson N, Romanos S, et al. A Roadmap for Developing Plasma Diagnostic and Prognostic Biomarkers of Cerebral Cavernous Angioma With Symptomatic Hemorrhage (CASH). Neurosurgery (2021) 88:686–97. doi: 10.1093/neuros/nyaa478
Keywords: cerebral cavernous malformations, vascular anomalies, endothelial cell homeostasis, inflammation, immune microenvironment
Citation: Tu T, Peng Z, Ren J and Zhang H (2022) Cerebral Cavernous Malformation: Immune and Inflammatory Perspectives. Front. Immunol. 13:922281. doi: 10.3389/fimmu.2022.922281
Received: 17 April 2022; Accepted: 18 May 2022;
Published: 30 June 2022.
Edited by:
Yujie Chen, Army Medical University, ChinaReviewed by:
Gang Chen, First Affiliated Hospital of Soochow University, ChinaJaesung Peter Choi, University of Technology Sydney, Australia
Copyright © 2022 Tu, Peng, Ren and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongqi Zhang, xwzhanghq@163.com
†These authors have contributed equally to this work and share first authorship